Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 43
Hírvilág
2011. AUGUSZTUS 15.


Gondolat
2011. AUGUSZTUS 15.

Csontvázakat is azonosított a professzor DNS profillal
A világon először használta szisztematikusan a DNS profilt tömegkatasztrófa áldozatainak azonosításához 1994-ben. A Debreceni Orvostudományi Egyetemen diplomázó, s 1974 óta Svájcban élő dr. Krompecher Tamás professzor, a svájci Lausanne-i Egyetem Igazságügyi Orvostani Intézetének korábbi igazgatója, aki előadásokat tart szerte a világon módszereiről, a hullamerevséggel kapcsolatos felfedezéseiről. A professzort a Debreceni Egyetem díszdoktorává avatták 2010 őszén.
Gondolat
2011. AUGUSZTUS 15.

Nem tükörkép hasonmások Az ikrek világa
Az iker-testvérek számát 125 millióra becsülik a világon. Statisztikák szerint minden 333 szülésre esik egy olyan, amikor egy terhesség után két baba jön világra.
Lege Artis Medicinae
2009. ÁPRILIS 01.

Cisztás fibrosis: felnőttellátás 20 év tapasztalattal
A cisztás fibrosis - mucoviscidosis - a leggyakoribb genetikai megbetegedés a kaukázusi népcsoportban. Autoszomális, recesszív öröklődésű, a tüdőt, hasnyálmirigyet, bőrt, a reproduktív szerveket, a máj- és a vékonybélműködést érinti. Az exokrin mirigyekben a csökkent kloridkiválasztás és a fokozott nátriumion-, vízvisszaszívás miatt vízben szegény, sűrű, viszkózus nyák képződik.


Lege Artis Medicinae
2009. MÁRCIUS 21.
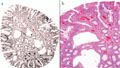
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.
Ideggyógyászati Szemle
2009. JANUÁR 20.

Genetikailag meghatározott neuromuscularis betegségek Magyarországon élõ roma családokban (angol nyelven)
A szerzők négy örökletesen meghatározott neuromuscularis kórkép klinikai és molekuláris genetikai aspektusait tenkintik át néhány magyarországi roma családban. Az autoszomális recesszíven öröklődő spinalis muscularis atrophia (SMA) csoportból nyolc, kaukázusi rasszhoz tartozó csecsemőben, kisdedben igazolták az SMA gén 7-8 exonalis deletióját, csupán két beteg tartozott a roma populációhoz. Hangsúlyozzuk, hogy a molekuláris genetikai lelet megegyezett a kaukázusi és roma populációban.
Ideggyógyászati Szemle
2008. DECEMBER 20.

Herediter nyomásérzékeny neuropathia gyermekkorban
A herediter nyomásérzékeny neuropathia autoszomális domináns öröklődésű kórkép, amit klinikailag rekurráló perifériás paresisek jellemeznek többnyire benignus lefolyással. Kiváltó okként gyakori a minor trauma vagy toxikus behatás. A perifériás ideg szövettani képe alapján „tomaculous” neuropathiának is nevezik, mivel a szegmentális de- és remyelinisatio a perifériás idegen hurkafűzérszerű képet okoz.
Lege Artis Medicinae
2008. MÁJUS 26.
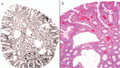
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.
Lege Artis Medicinae
2008. ÁPRILIS 22.

Hereditaer angioneuroticus oedema
A hereditaer angioneuroticus oedema autoszomális domináns öröklődésű. A közvetlen hozzátartozóknál az esetek körülbelül 85%-ában kimutatható, és a C1-INH gén mutációját hordozó betegek 98%-a tünetekkel is küzd.
Ideggyógyászati Szemle
2007. MÁJUS 20.

Congenitalis cataracta facialis dysmorphismus neuropathia szindróma Első magyarországi közlés
A congenitalis cataracta facialis dysmorphia neuropathia (CCFDN) szindróma (OMIM 604168) nemrég leírt autoszomális recesszív öröklődésű betegség, amely csaknem kizárólag az európai oláh cigányok endogám közösségére, a rudari csoportra korlátozódik.
1.
2.
3.
4.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
5.
1.
2.
Klinikai Onkológia
Hasnyálmirigyrák: az ESMO klinikai gyakorlati irányelve a diagnózishoz, kezeléshez, követéshez*
AUG 29.
3.
Klinikai Onkológia
Gyógyszerbiztonsági szemelvények – a múlt tanulságai és a jövő lehetőségei
AUG 29.
4.
5.