Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 43
Lege Artis Medicinae
2020. ÁPRILIS 18.
Digitális eszközökkel támogatott terápiatervezés folyamata a precíziós onkológiában
A molekuláris információ alapú személyre szabott precíziós orvoslás új mérföldkőhöz érkezett az onkológiában. Mostani tudásunk szerint hozzávetőleg 600 gén 6 millió mutációja hozható összefüggésbe a daganatok kialakulásával, és minden beteg esetében átlagosan egyszerre 3-4 ilyen „driver” gén mutációja van jelen. A molekuláris diagnosztika fejlődése ma már lehetővé teszi, hogy a rutinellátás részeként megismerjük a betegek daganatának részletes molekuláris profilját. Ennek klinikai relevanciája az, hogy ma már 125 célzott gyógyszer van forgalomban és további több száz hatóanyag érhető el klinikai vizsgálatokban. Ez azt eredményezi, hogy sokszor már első vonalban több törzskönyvezett terápiás lehetőség közül kell kiválasztani a beteg számára a legmegfelelőbbet a molekuláris információ alapján. Ehhez ma már egyre inkább komplex informatikai eszközökre, orvosi szoftverekre van szükség. Genetikusok, molekuláris biológusok, molekuláris patológusok és molekuláris farmakológusok már most napi szinten használnak bioinformatikai és interpretációs szoftvereket. De ma már klinikusok számára is online elérhetők a mesterséges intelligencia által támogatott digitális eszközök a terápia megtervezésére. A telemedicina eszközei, a videokonferencia megteremtette a feltételét annak, hogy online multidiszciplináris szakértői egyeztetések, „virtual molecular tumor board”-ok jöjjenek létre, amelyek segítségével minden orvos és beteg hozzáférhet a precíziós onkológia korszerű lehetőségeihez.



Ideggyógyászati Szemle Proceedings
2022. JÚNIUS 16.

Négy Niemann–Pick C esetünk, diagnosztikus és terápiás lehetőségek
42 éves nő. Újszülöttként icterus, hepatomegalia, intrahepaticus epeút-hypoplasia, cystoduodenostomia. Húszas évektől szellemi hanyatlás, ataxia, spasticus járás. Később myoclonusok, aluszékonyság, depresszió, akaratlan végtagmozgások, nyelészavar.
Ideggyógyászati Szemle
2021. NOVEMBER 30.
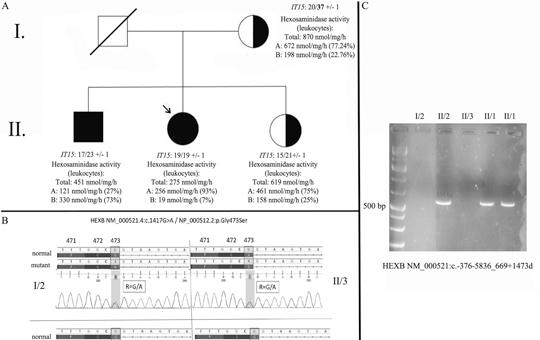
[Késői kezdetű Sandhoff-betegség atípusos jelentkezése: esetismertetés]
[Bevezetés – A Sandhoff-betegség egy olyan ritka, hereditaer GM2-gangliosidosis (autoszomális recesszív), amit a HEXB gén mutációja okoz. A hexózaminidáz (Hex) enzim β-alegységének károsodása miatt mind a Hex-A, mind a Hex-B izoformák működése zavart szenved. A betegség súlyossága, valamint a tünetek kezdete (infantilis vagy klasszikus, juvenilis, felnőttkori) a residualis enzimaktivitás függvénye. A késői kezdetű formát szerteágazó tünettan jellemzi. Jelen lehetnek többek között motoneuronbetegségre utaló eltérések, ataxia, tremor, dystonia, valamint pszichiátriai és neuropathiára utaló jegyek is. A 36 éves nőbeteg 9 éve tartó, progresszív, szimmetrikus alsó végtagi gyengeség miatt jelentkezett klinikánkon. A részletes neurológiai szakvizsgálat enyhe fokú szimmetrikus gyengeséget igazolt a csípőflexorokban, a többi izomcsoport megkíméltsége mellett. Mindkét oldalon a Patella-reflex renyhe volt. A laboratóriumi vizsgálatok releváns eltérést nem mutattak. A rutin elektro-encefalográfiás, valamint a koponya-MR-vizsgálatok a beteg panaszait magyarázó eltérést nem detektáltak. Az elektroneuronográfiás, valamint az elektromiográfiás vizsgálatokon szenzoros neuropathiának megfelelő eltérések látszottak. Az izombiopsziás minta elemzése kapcsán enyhe fokú neurogén károsodásra derült fény. A beteg öccse (32 éves) hasonló tüneteket mutat. Páciensünk részletes genetikai vizsgálata során két ismert patogén eltérést találtunk a HEXB génben, egy missense mutációt, valamint egy 15 008 bázispár hosszúságú deletiót (NM_000521.4:c.1417G>A; NM_000521:c.-376-5836_669+1473del; kettős heterozigóta állapot). A szegregációanalízis, valamint a családtagok hexózaminidáz-vizsgálata a késői kezdetű Sandhoff-betegség diagnózisát megerősítették. A jelen esetismertetés célja, hogy felhívja a figyelmet a késői kezdetű Sandhoff-betegség differenciáldiagnosztikai jelentőségére felnőttkorban kezdődő, proximális predominanciát mutató szimmetrikus alsó végtagi gyengeség esetén.]

Klinikai Onkológia
2021. FEBRUÁR 28.
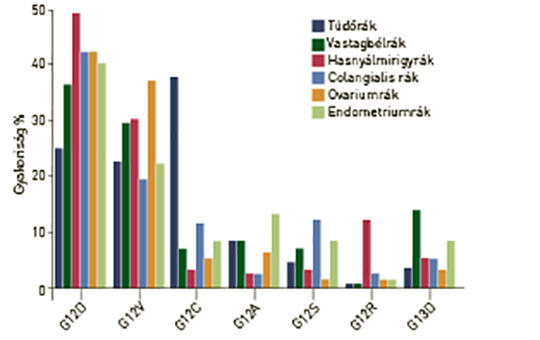
A (K)RAS-mutáció molekuláris epidemiológiája emberi daganatokban
A RAS onkogén mutációja a leggyakoribb génhiba emberi daganatokban, és a három családtag közül a K-RAS-é a leggyakoribb, amit az N-RAS követ. A tipikus K-RAS-mutáns daganatok a hasnyálmirigyrák, vastagbélrák és tüdő-adenocarcinoma, amelyekben a mutáns variáns allélok gyakorisága igen heterogén, aminek hátterében eltérő karcinogenezis áll. A RAS-mutáns daganatok genetikai sokszínűségét tovább fokozza, hogy a mutáns allél homo- vagy heterozigóta formában van-e jelen. A sokszínűség egy másik forrása az, hogy a különféle daganatokban a K-RAS-mutánsok esetében sajátos kísérő mutációs mintázatú altípusok lehetnek. Mindezeknek az a következménye, hogy a K-RAS-mutáns daganatok biológiai viselkedése és nagy valószínűséggel terápiás érzékenysége is nagyon heterogén lehet. A K-RAS-inhibitorok klinikai debütálásával ezeknek a kérdéseknek egyre nagyobb jelentősége lesz.
Ideggyógyászati Szemle
2020. MÁJUS 30.
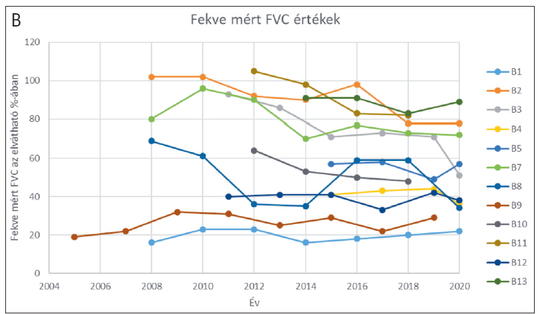
A késői kezdetű Pompe-kórban szenvedők enzimpótló kezelésének hosszú távú követése
A Pompe-kór (PD) egy ritka lizoszomális tárolási betegség, amit a GAA gén mutációja következtében kialakuló α-glükozidáz (GAA) enzim elégtelen működése okoz. Az enzimdeficientia a glikogén lizoszomális felszaporodásához vezet. A betegségnek két klinikai formája ismert, az újszülöttkori, valamint a késői forma. Jelenleg a betegség hátterében a GAA génnek közel 600 mutációja ismert. A kaukázusi populációban a késői forma hátterében a c.-32-13T>G mutáció a leggyakoribb, az allélfrekvencia közel 70%. A Pompe-kórt enzimpótló terápiával (ERT) tudjuk kezelni, kéthetente Myozyme infúzió adásával. Közleményünkben 13, több mint öt éve kezelt, késői kezdetű formában szenvedő beteg hosszú távú követését mutatjuk be. A leghosszabb követési idő 15 év volt. A kezelés eredményességének megítélésére évente mértük a 6 perces járótávolságot és a légzésfunkciót. Az adatok alapján a 6 perces járótávolság az enzimpótló kezelés indítása után körülbelül 3-4 évig javult, ezt követően az esetek többségében a megtett távolság csökkent. A több mint 10 éves követés után a kezdeti 6 perces járótávolsághoz képest romlást tapasztaltunk az esetek 77%-ában, javulást az esetek 23%-ában. A követés ideje alatt mindössze egyetlen beteg került kerekesszékbe. A légzésfunkció, különösen fekvő helyzetben hasonlóan alakult. A betegek terápiára adott válaszában nagy variabilitást figyeltünk meg, ami csak részben mutatott összefüggést a terápiás fehérje ellen termelődő antitestszinttel. Az ERT eredményessége jelentősen függött a betegséget okozó mutáció típusától, a betegség státuszától a kezelés kezdetekor, a beteg fizikai aktivitásától és táplálkozási szokásaitól. Az innovatív orphan gyógyszerekkel kezelt betegek hosszú távú követése kiemelkedően fontos ahhoz, hogy megismerjük a kezelés valós hasznát és a betegek igényeit.
Klinikai Onkológia
2020. ÁPRILIS 30.

A biológiai óra és a daganatok
Jelen összefoglalásban képet adunk a biológiai ritmusok közül a cirkadián ritmusról, annak szabályozásáról és a tumorgenezissel való kapcsolatáról. Cirkadián ritmusnak nevezzük azt a biokémiai, fiziológiai folyamatokban fellépő nagyjából 24 órás ciklust, amely az egysejtűektől a gerincesekig megtalálható. Ez a biológiai ritmus az endogén belső óráink és a fény mint fő „Zeitgeber” szinkronizációjának eredménye. Az emlősökben a hypothalamus területén elhelyezkedő nucleus suprachiasmaticus (SCN) tekinthető a szervezet „főórájának”, ami felelős a különböző szervrendszerekben található perifériás órák összehangolásáért. A cirkadián ritmus szabályozása az úgynevezett „circadian locomotor output cycles kaput”, a CLOCK géneknek a feladata. A CLOCK gének más effektor génekre hatva, a fehérjeszintézis diurnalis ritmusát szabályozzák. A cirkadián ritmus celluláris mechanizmusainak feltárását 2017-ben Nobel-díjjal jutalmazták. Egyre több adat bizonyítja a CLOCK gének és a daganatok kialakulása közötti összefüggést. Több tanulmány kapcsolatot mutat a váltott műszakban történő munkavégzés és az emlő-, valamint prosztatarák kialakulása között, valamint egyes cirkadián ritmust szabályozó gének mutációja és a tumoros elváltozás kifejlődése között. Egyre több adat utal a tumormetabolizmus és a CLOCK gének, azok szabályozása közötti kapcsolatra is. Mindezen adatok alapján a cirkadián ritmus, így a napszakok figyelembevétele a daganatterápiás kezelés során indokolttá válhat.
Ideggyógyászati Szemle
2019. JANUÁR 30.
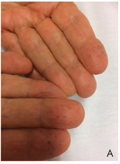
Multiplex ischaemiás stroke Osler-Rendu-Weber-kórban
A hereditaer haemorrhagiás teleangiectasia (HHT, Osler-Rendu-Weber-kór) olyan autoszomális domináns módon öröklődő, több lehetséges gén mutációja által okozott megbetegedés, amit az arteriovenosus rendszer több szervben is megjelenő malformációja jellemez. A klinikai diagnózist a Curaçao-kritériumok [1. spontán, visszatérő epistaxis; 2. karakterisztikus lokalizációban elhelyezkedő teleangiectasiák (ajak, szájüreg, orr, ujjak); 3. visceralis laesiók (gastrointestinalis, pulmonalis, cerebralis, spinalis); 4. elsőfokú érintett családtag] alapján állíthatjuk fel. A jelen közlemény célja Magyarországon elsőként egy multiplex ischaemiás stroke-kal társuló, genetikailag igazolt HHT-s eset ismertetése.
Lege Artis Medicinae
2016. DECEMBER 18.
A családorvos feladatai a Wilson-kór miatt szükséges májtranszplantáció kapcsán
A Wilson-kór a 13-as kromoszómán lévő ATP7B gén mutációja következtében kialakuló, autoszomális recesszív módon öröklődő, rézfelhalmozódással járó betegség.
Klinikum
2016. OKTÓBER 11.

T-sejt-alcsoportok és jelentőségük autoimmun és reumatológiai betegségekben - A Figyelő 2015;2
A T-sejtek heterogén sejtpopuláció, tagjai sejt felszíni markereik, transzkripciós faktoraik és citokintermelésük alapján egymástól markánsan elhatárolható alcsoportokra oszthatóak.
Ideggyógyászati Szemle
2016. MÁJUS 30.
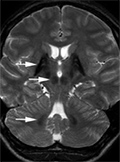
Genetikailag meghatározott, agyi vasfelhalmozódással és neurodegenerációval járó kórképek
A kóros központi idegrendszeri vasfelhalmozódással, progresszív, jellemzően bazális ganglion károsodásra utaló tünetekkel járó, ritka, genetikailag meghatározott betegségcsoportot összefoglaló néven NBIA-nak (neurodegeneration with brain iron accumulation, neurodegeneráció agyi vasfelhalmozódással) nevezik.
1.
2.
3.
4.
5.
Egészségpolitika
Hadiállapotként kezeli és így is reagál a kormány az egészségügy „rendezésére”
2024 JÚN 18.
1.
2.
Mesterséges intelligencia
A legújabb, MI által asszisztált robotsebészet megérkezett Magyarországra
2024 SZEPT 27.
3.
Orvoslás és társadalom
Különleges nap halmozottan sérült gyermekeket nevelő szülők számára a Bükki Nemzeti Parkban
2024 SZEPT 27.
4.
5.