Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 43
Lege Artis Medicinae
2009. MÁRCIUS 21.
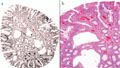
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.



Ideggyógyászati Szemle
2008. DECEMBER 20.
A dravet-szindróma klinikai és genetikai diagnosztikájáról húsz esetünk kapcsán
A Dravet-szindrómát vagy más néven súlyos csecsemőkori myoclonus epilepsziát az addig minden szempontból jól fejlődő csecsemő esetében az első életévben fellépő, eleinte többnyire lázas, majd láztalan, elhúzódó, generalizált vagy féloldali, általában változó oldali, clonusos vagy tónusos-clonusos rohamok jellemzik, amelyekhez később myoclonusok, atípusos absence-ok, komplex parciális rohamok társulnak.
Lege Artis Medicinae
2008. MÁJUS 26.
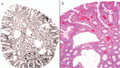
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.
Ideggyógyászati Szemle
2007. JÚLIUS 30.
DYT1-pozitív generalizált dystonia: Egy testvérpár esettanulmánya
A korai kezdetű generalizált dystoniákra jellemző a széles klinikai fenotípusspektrum és a szűk terápiás lehetőségek. A mozgászavarok ilyen formáinak több mint 50%-ában a DYT1 gén mutációja felelős a kórkép kialakulásáért.
Ideggyógyászati Szemle
2007. MÁJUS 20.

Congenitalis cataracta facialis dysmorphismus neuropathia szindróma Első magyarországi közlés
A congenitalis cataracta facialis dysmorphia neuropathia (CCFDN) szindróma (OMIM 604168) nemrég leírt autoszomális recesszív öröklődésű betegség, amely csaknem kizárólag az európai oláh cigányok endogám közösségére, a rudari csoportra korlátozódik.
Lege Artis Medicinae
2006. OKTÓBER 18.
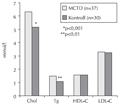
A paraoxonázaktivitás vizsgálata kevert kötőszöveti betegségben
A kevert kötőszöveti betegség több szervet érintő gyulladásos autoimmun kórkép. Az immunoinflammatorikus folyamatok jelentős szerepet játszanak az atherosclerosis patogenezisében. Kevert kötőszöveti betegségben még nem vizsgálták a gyulladásos paraméterek és az atherosclerosis kapcsolatát.
Magyar Immunológia
2006. MÁRCIUS 20.
A pulmonalis artériás hipertenzió kórlefolyása, klinikai és immunszerológiai jellemzői kevert kötőszöveti betegségben
Kevert kötőszöveti betegséghez (mixed connective tissue disease, MCTD) társuló pulmonalis artériás hipertenzióban szenvedő betegek kórlefolyását, a túlélés valószínűségét, és az immunszerológiai eltéréseket vizsgáltuk. Az eredményeket összevetettük a pulmonalis artériás hipertenzió nélküli MCTD-s betegek értékeivel.
Magyar Radiológia
2005. DECEMBER 10.

A férfiak emlőrákja
A férfiak emlőrákja semmilyen vonatkozásban nem kapja meg azt a figyelmet, amelyet jellegének következtében megérdemelne. Hiányoznak a diagnosztikai és terápiás protokollok, ezek létrehozásához országos centrumra és nemzetközi összefogásra volna szükség.
Magyar Radiológia
2005. FEBRUÁR 15.
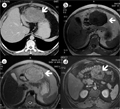
Gastrointestinalis stromalis tumorok
A gastrointestinalis stromalis tumorok az emésztőrendszer leggyakoribb mesenchymalis daganatai. Differenciációs potenciáljuk széles variációt mutat. A gastrointestinalis stromalis tumorokat a c-kit gén (transzmembrán tirozinkináz) mutációja jellemzi, amely a KIT protein expressziójával igazolható. Ez a fehérje tirozinkináz-aktivitással rendelkezik. A KIT-expresszió (CD117) immunhisztokémiai módszerekkel történő kimutatásával lehetséges a gastrointestinalis stromalis tumorokat az egyéb mesenchymalis daganatoktól - a leiomyomától, leiomyosarcomától, leiomyoblastomától és a schwannomától - elkülöníteni. A patológiailag igazolt gastrointestinalis stromalis tumorok alkalmasnak tekinthetők a molekuláris szinten ható KIT-inhibitor- terápiára.
Ideggyógyászati Szemle
2004. JÚNIUS 10.

Az epilepszia genetikája
Összefoglaló közleményünkben olyan epilepsziaszindrómákat ismertetünk, amelyeknek oka egy-egy gén mutációja. Ezeknek a betegségeknek a zöme a feszültség- és ligandfüggő ioncsatornák működési zavarára vezethetők vissza. A herediter epilepsziák közül leginkább az autoszomális domináns öröklődésmenetű parciális epilepsziákat ismerjük; az autoszomális domináns öröklődésű frontálislebeny-epilepsziát az acetil-kolin-receptor α-4 és β-2 alegységeit kódoló gének mutációja, az újszülöttkori görcsöket a káliumcsatorna-alegységeket (KCNQ2 és KCNQ3) megváltoztató génmutációk, a familiáris temporolateralis epilepsziát egy tumorszuppresszor gén mutációja okozza.
1.
2.
3.
4.
5.
Egészségpolitika
Hadiállapotként kezeli és így is reagál a kormány az egészségügy „rendezésére”
2024 JÚN 18.
1.
2.
Mesterséges intelligencia
A legújabb, MI által asszisztált robotsebészet megérkezett Magyarországra
2024 SZEPT 27.
3.
Orvoslás és társadalom
Különleges nap halmozottan sérült gyermekeket nevelő szülők számára a Bükki Nemzeti Parkban
2024 SZEPT 27.
4.
5.