Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 47
Hírvilág
2023. NOVEMBER 29.

Egyéves az önkéntes újszülöttkori SMA-szűrés program
A gerincvelő-eredetű izomsorvadással (SMA) született gyermekeket a betegséget módosító gyógyszeres terápiával egészen 2018-ig nem tudták kezelni, emiatt a legsúlyosabb formával érintettek sokszor az egyéves kort sem élték meg, melynek oka többnyire a súlyos légzési elégtelenség volt.


Hírvilág
2023. JÚNIUS 29.

Az EU-ban évente 4000 gyermek életét mentheti meg a kötelező újszülöttkori szűrés
2023. június 28-án ünnepeljük a Nemzetközi Újszülöttkori Szűrés Világnapját, melynek célja felhívni a figyelmet az újszülöttkori szűrés fontosságára és a korai diagnózis jelentőségére. A szűrésnek köszönhetően Európában évente mintegy 4,2 millió újszülöttet tesztelhetnek veleszületett fejlődési rendellenességre és anyagcsere-betegségre.
Ideggyógyászati Szemle Proceedings
2022. JÚNIUS 16.

Újszülöttkori koponyasérülések
A szerzők előadásukban ismertetik a Debreceni Egyetemen az elmúlt húsz évben kezelt újszülöttkori koponyasérülést szenvedett gyermekek eseteit. A szülés kapcsán a leggyakoribb sérülés az ún. pingponglabdatörés: fedett sérülés, ami általában a parietalis tájékon kerül észlelésre.


Ideggyógyászati Szemle Proceedings
2022. JÚNIUS 16.

Az időfaktor szerepe a betegségmódosító terápiák hatékonyságában spinalis muscularis atrophiában szenvedő gyermekeknél
A veleszületett gerincvelői izomsorvadás gyógyíthatatlan genetikai betegség. Legsúlyosabb formájában a betegek 75%-át elveszítettük 20 hónapos korukig. A 21. század áttörése a betegségmódosító gyógyszerek forgalomba kerülése. Jelenleg az SMN2-gén funkciójának javítását célzó intrathecalis nusinersen és per os risdiplam, valamint a hiányzó SMN1-gént pótló intravénás készítmény áll rendelkezésre. Hatékonyságuk függ a betegek terápiakezdeti életkorától, ezért kulcsfontosságú a betegség korai felismerése.
Klinikum
2021. JÚNIUS 09.
A vérplazma összetevői, azok élettani szerepe, és gyógyászati felhasználása
A vér folyékony kötőszövetünk komplex bonyolult és összetett funkcióval. Alkotórészei a sejtes elemek (vörösvérsejtek, fehérvérsejtek, vérlemezkék), és az in vivo működésükhöz szükséges közeg a vér 55%-át kitevő plazma. Ebben a sejtek szabadon áramlanak, lehetővé teszi, hogy azok minden szervhez eljuthassanak az érpályán keresztül és kifejthessék élettani hatásukat.
Ideggyógyászati Szemle
2021. MÁRCIUS 30.
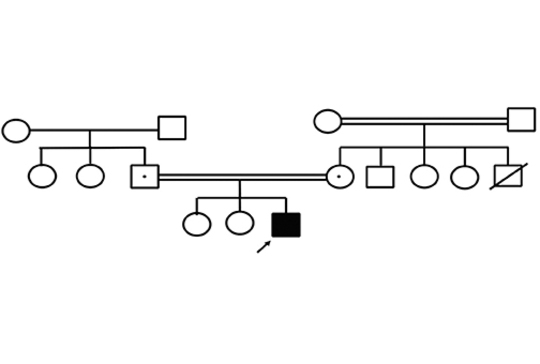
[Új patogén mutáció következtében kialakuló karnitin-palmitoil-transzferáz II-hiánnyal magyarázható ismétlődő rhabdomyolysis]
[A karnitin-palmitoil-transzferáz II- (CPT II-) hiány egy autoszomálisan öröklődő anyagcsere-rendellenesség, amelyben a hosszú láncú zsírsavak β-oxidációja hiányos. A klinikai megjelenés különféle formában lehetséges; súlyos formájában jelentkezik újszülöttkorban és infantilis időszakban, míg iskolás- és serdülőkorban a kevésbé súlyos myopathiás formában jelentkezik. Bár a rhabdomyolysisrohamok súlyossága változó, a klinikai lefolyást esetenként akut veseelégtelenség komplikálhatja. Az acilkarnitin-elemzés segíthet a CPT II diagnosztizálásában, de az eredmény normalitása nem jelzi a betegség hiányát. Erős gyanú esetén genetikai elemzést kell végezni. Ebben a tanulmányban egy 15 éves fiú beteg esetét mutatjuk be, akinek két, fertőzés, illetve éhezés által kiváltott rhabdomyolysisrohama volt. Az acilkarnitin-elemzés eredménye normális volt, a kórtörténet értékelésénél figyelembe vettük a CPT II-hiányt, és kimutattuk a CPT II gén c.137A> G (p.Gln46Arg) új patogén homozigóta mutációját. A CPT II-hiány a metabolikus rhabdomyolysis egyik leggyakoribb oka az ismétlődő rhabdomyolysisepizódoktól szenvedő betegek esetén.]
Ideggyógyászati Szemle
2021. MÁRCIUS 30.
[Lehetséges genotípus-fenotípus korrelációk C típusú Niemann–Pick-kórban és a betegek miglustatkezelése]
[A C típusú Niemann–Pick-kór károsodott intracelluláris koleszterintranszport miatt kialakuló, ritka lizoszomális tárolási betegség. Az autoszomális recesszív betegséget az NPC1 vagy NPC2 gén mutációi okozzák. Értékeltük kora újszülöttkori, C típusú Niemann–Pick-kórral diagnosztizált betegeink klinikai és laboratóriumi tüneteit, a genotípusuk és a fenotípusuk közötti korrelációt és a miglustatkezelésre adott válaszukat. Tanulmányunkban bemutatjuk négy, C típusú Niemann–Pick-kórral kora újszülöttkorban diagnosztizált beteg esetét. Betegeink közös tünete volt a hepato- és splenomegalia, a cholestasis és a mozgásfejlődés visszamaradása. Az 1-es és a 2-es beteg (ikrek) az NPC1 gén c.2776G>A p.(Ala926Thr) homozigóta mutációjával és súlyos fokú tüdőérintettséggel bírt. A tüdőérintettség, ami a szakirodalom szerint általában az NPC2 gén mutációjával jár együtt, betegeinkben olyan súlyos fokú volt, ami korai halálukat eredményezte. A 3-as és 4-es beteg az NPC1 gén c.2972del p.(Gln991Argfs*6) mutációjával és az NPC2 gén homozigóta c.133C>T p.(Gln45*) mutációjával bír. Ennél a két betegnél miglustatkezelés hatására a neurológiai tünetek javulását figyeltük meg. Ikerpár betegeinknél súlyos fokú tüdőérintettséget figyeltünk meg. A négy közül kettő, kora újszülöttkori, C típusú Niemann–Pick-kórral diagnosztizált betegünk neurológiai tünetei a miglustatkezelés hatására javultak.]
Egészségpolitika
2020. AUGUSZTUS 18.
Gyermekpszichiátria - sürgős beavatkozásra van szükség!
A nemzetközi vizsgálatok szerint a fogyasztói társadalmakban minden ötödik gyermek- és serdülőkorú küzd fejlődési, érzelmi vagy viselkedési problémákkal, és tízből egynél mentális betegség diagnosztizálható. Magyar adatok szerint a 14 év alattiak az egészséges életév veszteség második leggyakoribb oka a mentális betegségek és viselkedés zavarok, az újszülöttkori megbetegedések után. A WHO 2020-ra a gyermekpszichiátriai megbetegedések megduplázódását, s általában is a mentális betegségek népbetegséggé válását jelezték, ami úgy tűnik igazolódott. Ehhez képest ma öt olyan megye van, ahol egyetlen szakember sincs, gyerekek ezrei vannak ellátatlanul, sokszor még diagnózisig sem jutnak. Vészhelyzet van.
Ideggyógyászati Szemle
2020. JÚLIUS 30.
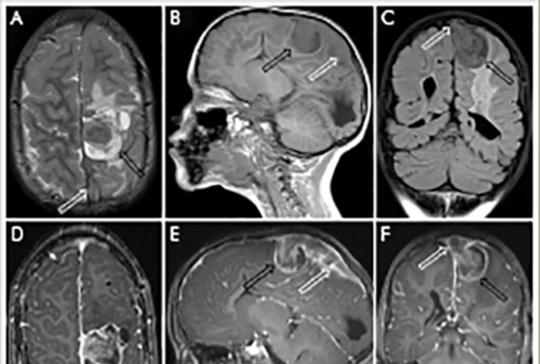
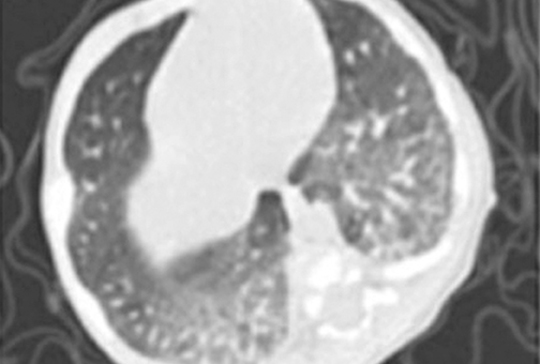
[Extraskeletalis, intraduralis, nem metasztatikus Ewing-sarcoma]
[Intracranialis lokalizációjú Ewing-sarcoma nagyon ritkán fordul elő. Egy négyéves fiúgyermek klinikai és képalkotó vizsgálatainak jellegzetességeit ismertetjük. Koraszülött volt, intraventricularis vérzés szövődményeként kialakult posthaemorrhagiás hydrocephalus miatt ventriculoperitonealis sönt beültetésen esett át újszülöttkorában. Rendszeres gondozás során nem észleltük söntvezetési zavar vagy emelkedett intracranialis nyomás tüneteit. Nyolc hónapos korában készült utolsó képalkotó vizsgálata. Négyéves korában ismétlődő hányás, fokális epilepsziás rohamok kezdődtek. Koponya-MR-vizsgálata bal oldali frontoparietalis, a sinus sagittalis superiorba betörő térfoglaló folyamatot mutatott. Craniotomia során a tumor teljes eltávolításra került. A tumorszövet hisztológiai vizsgálata igazolta a kis, kék, kerek sejtes daganatot. A Ewing-sarcoma diagnózisát az EWSR1-géntranszlokáció kimutatása FISH-módszerrel megerősítette. A staging vizsgálatokkal metasztázis nem volt kimutatható. A beteg az EuroEwing99 protokoll szerint kapta meg kezelését. 10 év telt el a diagnózis és a műtét óta, jelenleg is tumor- és rohammentes, életminősége jó.]
Ideggyógyászati Szemle
2020. MÁJUS 30.
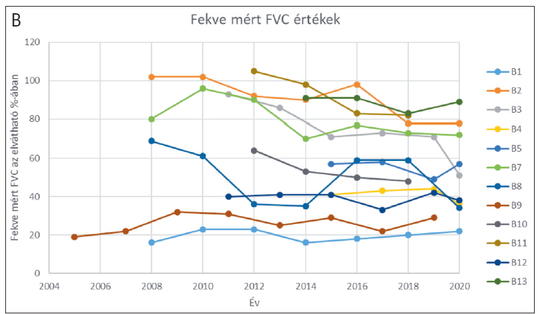
A késői kezdetű Pompe-kórban szenvedők enzimpótló kezelésének hosszú távú követése
A Pompe-kór (PD) egy ritka lizoszomális tárolási betegség, amit a GAA gén mutációja következtében kialakuló α-glükozidáz (GAA) enzim elégtelen működése okoz. Az enzimdeficientia a glikogén lizoszomális felszaporodásához vezet. A betegségnek két klinikai formája ismert, az újszülöttkori, valamint a késői forma. Jelenleg a betegség hátterében a GAA génnek közel 600 mutációja ismert. A kaukázusi populációban a késői forma hátterében a c.-32-13T>G mutáció a leggyakoribb, az allélfrekvencia közel 70%. A Pompe-kórt enzimpótló terápiával (ERT) tudjuk kezelni, kéthetente Myozyme infúzió adásával. Közleményünkben 13, több mint öt éve kezelt, késői kezdetű formában szenvedő beteg hosszú távú követését mutatjuk be. A leghosszabb követési idő 15 év volt. A kezelés eredményességének megítélésére évente mértük a 6 perces járótávolságot és a légzésfunkciót. Az adatok alapján a 6 perces járótávolság az enzimpótló kezelés indítása után körülbelül 3-4 évig javult, ezt követően az esetek többségében a megtett távolság csökkent. A több mint 10 éves követés után a kezdeti 6 perces járótávolsághoz képest romlást tapasztaltunk az esetek 77%-ában, javulást az esetek 23%-ában. A követés ideje alatt mindössze egyetlen beteg került kerekesszékbe. A légzésfunkció, különösen fekvő helyzetben hasonlóan alakult. A betegek terápiára adott válaszában nagy variabilitást figyeltünk meg, ami csak részben mutatott összefüggést a terápiás fehérje ellen termelődő antitestszinttel. Az ERT eredményessége jelentősen függött a betegséget okozó mutáció típusától, a betegség státuszától a kezelés kezdetekor, a beteg fizikai aktivitásától és táplálkozási szokásaitól. Az innovatív orphan gyógyszerekkel kezelt betegek hosszú távú követése kiemelkedően fontos ahhoz, hogy megismerjük a kezelés valós hasznát és a betegek igényeit.
1.
2.
3.
4.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
5.
1.
2.
Klinikai Onkológia
A rosszindulatú daganatok fenotípusának plaszticitása és az immunogén mimikri
AUG 29.
3.
Klinikai Onkológia
A szarkopénia mérése komputertomográfiával és jelentősége az onkológiai betegeknél
AUG 29.
4.
5.