Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 40
Lege Artis Medicinae
2006. SZEPTEMBER 15.

Emlőrák az onkológiai gyakorlatban A megelőzéstől a gondozásig
Az emlőrák a fejlett országok leggyakoribb női daganatos betegsége. Kialakulásáért legtöbbször különféle hormonok tehetők felelőssé, és csak az összes eset 10%-a veleszületett génmutáció következménye.



Magyar Immunológia
2006. MÁRCIUS 20.
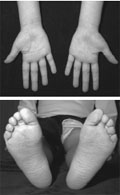
Familiáris autoinflammatoricus szindrómák
A látszólag ok nélküli, nagyfokú gyulladással zajló, ritka örökletes betegségcsoport, az úgynevezett familiáris autoinflammatoricus szindrómák gyermekkortól induló ritka, de egész életen át tartó tünetegyüttesek.
Magyar Radiológia
2005. OKTÓBER 10.
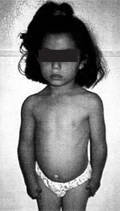
Du Pan-féle acromesomelicus dysplasia
A porc eredetű morfogenikus fehérje (CDMP1) génmutációi számos rendellenességért felelõsek, az intrauterin elhalástól az egészen enyhe anomáliákig, mint a C típusú brachydactylia. A CDMP1 mutációnak két hasonló megjelenésű súlyos formája van, a du Pan- és a Hunter-Thompson-szindróma.
Ideggyógyászati Szemle
2005. JÚLIUS 10.

Frontotemporalis dementia, II. rész - Differenciáldiagnózis, genetika, molekuláris patomechanizmus és patológia
A szerzők három részből álló összefoglaló közleményükben a frontotemporalis dementia történetét, előfordulását, klinikai megjelenési formáit, a megkülönböztető kórismét, genetikáját, molekuláris patomechanizmusát, patológiáját, valamint terápiáját tekintik át.
Magyar Radiológia
2004. OKTÓBER 20.
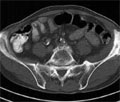
Képalkotó vizsgálatok a malignus lymphomák diagnosztikájában és követésében
Mai ismereteink szerint egyes vírusfertőzéseknek, génmutációknak és az immunszuppressziónak szerepe lehet a malignus lymphomák kialakulásában. A diagnózis idején észlelhető klinikai stádium fontos tényező a betegség prognózisában. A nodalis és extranodalis státus felmérését standardizált képalkotó módszerekkel végezzük. A leggyakoribb extranodalis manifesztációk a csontvelőben,
Ideggyógyászati Szemle
2004. JÚNIUS 10.

Az epilepszia genetikája
Összefoglaló közleményünkben olyan epilepsziaszindrómákat ismertetünk, amelyeknek oka egy-egy gén mutációja. Ezeknek a betegségeknek a zöme a feszültség- és ligandfüggő ioncsatornák működési zavarára vezethetők vissza. A herediter epilepsziák közül leginkább az autoszomális domináns öröklődésmenetű parciális epilepsziákat ismerjük; az autoszomális domináns öröklődésű frontálislebeny-epilepsziát az acetil-kolin-receptor α-4 és β-2 alegységeit kódoló gének mutációja, az újszülöttkori görcsöket a káliumcsatorna-alegységeket (KCNQ2 és KCNQ3) megváltoztató génmutációk, a familiáris temporolateralis epilepsziát egy tumorszuppresszor gén mutációja okozza.
Ideggyógyászati Szemle
2002. DECEMBER 20.
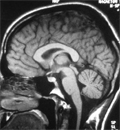
Friedreich-ataxia - diagnózis egy évtized után. Az örökletes spinocerebellaris ataxiák elkülönítése
Az örökletes spinocerebellaris ataxiák klinikai diagnózisa az egymással átfedésben lévő fenotípusok miatt nehéz. A szerzők röviden áttekintik a differenciáldiagnosztikai szempontból fontos örökletes ataxiával járó tünetegyütteseket, részletesebben tárgyalva a leggyakrabban előfordulót, a Friedreich-ataxiát.
Lege Artis Medicinae
2000. FEBRUÁR 01.

A mellékvesekéreg csökkent működésében manifesztálódó juvenilis haemochromatosis
A juvenilis haemochromatosis ritka, autoszomális recesszív módon öröklődő, fiatal felnőttkorban jelentkező megbetegedés, amelyet az időskorban manifesztálódó, szintén genetikusan determinált formával, az úgynevezett adult típusúval egyező módon a vas korlátlan felszívódása és a parenchymás szervekben történő lerakódása jellemez. Főként a máj, a lép, a pancreas, a szív és a bőr érintett. Ritkán a hypophysis is károsodik, ilyenkor szekunder módon gonadalis hipofunkció alakul ki. ESETISMERTETÉS - Bemutatott betegünk esetében primer adrenalis hipofunkció hátterében igazoltuk a juvenilis haemochromatosis fennállását. A szív érintettsége már a diagnózis megállapításakor kimutatható volt, míg a máj szövettani vizsgálatával fokozott vastárolás nem volt igazolható, szerkezete ép volt. Genetikai analízis segítségével megállapítottuk, hogy betegünk nem hordozza a genetikai haemochromatosis HFE-génjének C282Y mutációját, míg a H63D-mutációra nézve heterozigótának bizonyult. Hormonpótló kezelés és rendszeres vérlebocsátás mellett a beteg panaszmentes. KÖVETKEZTETÉS - Az adult típusú haemochromatosis szempontjából kórjelző C282Y mutáció hiánya alapján valószínűsíthető a juvenilis haemochromatosis kórképe, amelynek hát terében egy másik gén eltérése feltételezhető.
Lege Artis Medicinae
1991. AUGUSZTUS 28.

Germinális mutációk Magyarországi felügyelelte
A szerzők a Germinális Mutációk Magyarországi Felügyelete tízéves tevékenységének eredményeiről számolnak be. A felügyelet célja a környezeti hatásokra létrejövő csíra sejt-mutációk veleszületett rendellenességeket okozó hatásainak epidemiológiai elemzése volt. Az epidemiológiai megfigyelésre alkalmas, úgynevezett indikátor rendellenességek három csoportját különítették el, melyek születéskori gyakoriságát 10 éven keresztül folyamatosan észlelték. Indikátor rendellenességek voltak: (1) Domináns öröklődést jelző fejlődési anomáliák, melyeket a dominánsan átörökíthető germinális génmutációk indikátorának tekintettek. A 15 rendellenesség születéskori gyakorisága a vizsgált időszakban (1980– 1989) 3,8/10 000 összes születés volt. A sporadikus esetek aránya (90%) alapján így 31/ 100 000 összes születés mutációs gyakoriságot figyeltek meg, mely szignifikáns változást a vizsgált periódusban nem mutatott. (2) Down-syndroma, melyet a számbeli germinális chromosoma-mutációk indikátoraként alkalmaztak. Születéskori prevalenciája 11,7/10 000 összes születés volt a vizsgált időszakban. Az összes eset csaknem 100%-a új mutáció, így az új mutációk gyakorisága 110/100 000 összes születés volt. (3) Nem azonosítható többszörös fejlődési rendellenességek csoportja, melyek epidemiológiai elemzése során 1985-ben erősen szignifikáns növekedés volt kimutatható a germinális mutációk gyakoriságában. Ez egy évvel a csernobili atomerőmű-szerencsétlenség előtt következett be, azt követően pedig nem volt lényeges változás az indikátor rendellenességek gyakoriságában. A lehetséges okok kutatása folyamatban van.
Ideggyógyászati Szemle
1985. AUGUSZTUS 01.

Neurocutan kórképek - sclerosis tuberosa és neurofibromatosis - klinikai genetikai aspektusai, HLA-antigén vizsgálatok és computer tomographiás diagnosztikája
A szerzők 5 sclerosis tuberosás (ST) családban végeztek HLA tipizálást, a HLA-A w31 antigén szignifikánsan gyakoribb előfordulását igazolták, a B an tigéncsoport érintettsége nélkül. 4 családban autosomalis domináns, 1 családban pedig első génmutációs eredet igazolódott, a családtagok kültakarójának UV fényben történő vizsgálata, a depigmentált foltok detektálható fluoreszcencia jelensége alapján. 6 neurofibromatosisos (Nf) eset közül 4 volt forme fruste alak, az öröklődésmenet 4 családban autosomalis domináns, 1 családban első génmutációs eredet volt bizonyítható. A szerzők a computer tomografia diagnosztikus értékét is tárgyalják a fenti kórképek esetében.
1.
2.
3.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
4.
5.
1.
2.
Klinikai Onkológia
A rosszindulatú daganatok fenotípusának plaszticitása és az immunogén mimikri
AUG 29.
3.
Klinikai Onkológia
A szarkopénia mérése komputertomográfiával és jelentősége az onkológiai betegeknél
AUG 29.
4.
5.