Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 58
Ideggyógyászati Szemle
2021. NOVEMBER 30.
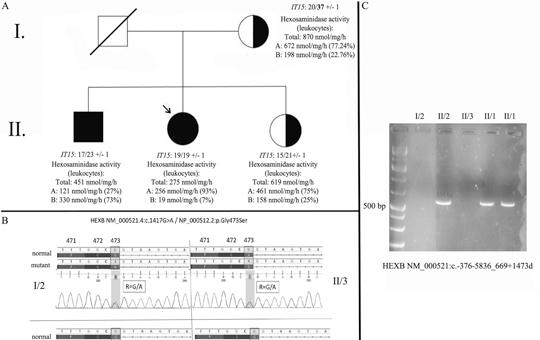
[Késői kezdetű Sandhoff-betegség atípusos jelentkezése: esetismertetés]
[Bevezetés – A Sandhoff-betegség egy olyan ritka, hereditaer GM2-gangliosidosis (autoszomális recesszív), amit a HEXB gén mutációja okoz. A hexózaminidáz (Hex) enzim β-alegységének károsodása miatt mind a Hex-A, mind a Hex-B izoformák működése zavart szenved. A betegség súlyossága, valamint a tünetek kezdete (infantilis vagy klasszikus, juvenilis, felnőttkori) a residualis enzimaktivitás függvénye. A késői kezdetű formát szerteágazó tünettan jellemzi. Jelen lehetnek többek között motoneuronbetegségre utaló eltérések, ataxia, tremor, dystonia, valamint pszichiátriai és neuropathiára utaló jegyek is. A 36 éves nőbeteg 9 éve tartó, progresszív, szimmetrikus alsó végtagi gyengeség miatt jelentkezett klinikánkon. A részletes neurológiai szakvizsgálat enyhe fokú szimmetrikus gyengeséget igazolt a csípőflexorokban, a többi izomcsoport megkíméltsége mellett. Mindkét oldalon a Patella-reflex renyhe volt. A laboratóriumi vizsgálatok releváns eltérést nem mutattak. A rutin elektro-encefalográfiás, valamint a koponya-MR-vizsgálatok a beteg panaszait magyarázó eltérést nem detektáltak. Az elektroneuronográfiás, valamint az elektromiográfiás vizsgálatokon szenzoros neuropathiának megfelelő eltérések látszottak. Az izombiopsziás minta elemzése kapcsán enyhe fokú neurogén károsodásra derült fény. A beteg öccse (32 éves) hasonló tüneteket mutat. Páciensünk részletes genetikai vizsgálata során két ismert patogén eltérést találtunk a HEXB génben, egy missense mutációt, valamint egy 15 008 bázispár hosszúságú deletiót (NM_000521.4:c.1417G>A; NM_000521:c.-376-5836_669+1473del; kettős heterozigóta állapot). A szegregációanalízis, valamint a családtagok hexózaminidáz-vizsgálata a késői kezdetű Sandhoff-betegség diagnózisát megerősítették. A jelen esetismertetés célja, hogy felhívja a figyelmet a késői kezdetű Sandhoff-betegség differenciáldiagnosztikai jelentőségére felnőttkorban kezdődő, proximális predominanciát mutató szimmetrikus alsó végtagi gyengeség esetén.]



Ideggyógyászati Szemle
2021. MÁRCIUS 30.
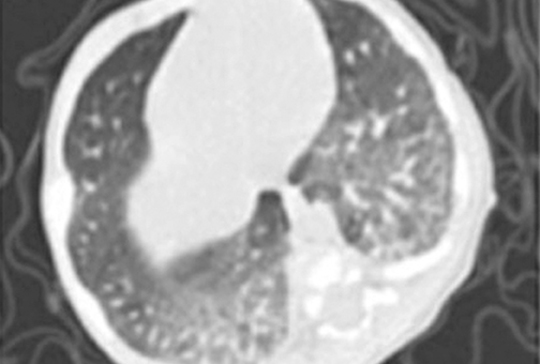
[Lehetséges genotípus-fenotípus korrelációk C típusú Niemann–Pick-kórban és a betegek miglustatkezelése]
[A C típusú Niemann–Pick-kór károsodott intracelluláris koleszterintranszport miatt kialakuló, ritka lizoszomális tárolási betegség. Az autoszomális recesszív betegséget az NPC1 vagy NPC2 gén mutációi okozzák. Értékeltük kora újszülöttkori, C típusú Niemann–Pick-kórral diagnosztizált betegeink klinikai és laboratóriumi tüneteit, a genotípusuk és a fenotípusuk közötti korrelációt és a miglustatkezelésre adott válaszukat. Tanulmányunkban bemutatjuk négy, C típusú Niemann–Pick-kórral kora újszülöttkorban diagnosztizált beteg esetét. Betegeink közös tünete volt a hepato- és splenomegalia, a cholestasis és a mozgásfejlődés visszamaradása. Az 1-es és a 2-es beteg (ikrek) az NPC1 gén c.2776G>A p.(Ala926Thr) homozigóta mutációjával és súlyos fokú tüdőérintettséggel bírt. A tüdőérintettség, ami a szakirodalom szerint általában az NPC2 gén mutációjával jár együtt, betegeinkben olyan súlyos fokú volt, ami korai halálukat eredményezte. A 3-as és 4-es beteg az NPC1 gén c.2972del p.(Gln991Argfs*6) mutációjával és az NPC2 gén homozigóta c.133C>T p.(Gln45*) mutációjával bír. Ennél a két betegnél miglustatkezelés hatására a neurológiai tünetek javulását figyeltük meg. Ikerpár betegeinknél súlyos fokú tüdőérintettséget figyeltünk meg. A négy közül kettő, kora újszülöttkori, C típusú Niemann–Pick-kórral diagnosztizált betegünk neurológiai tünetei a miglustatkezelés hatására javultak.]

Ideggyógyászati Szemle
2020. SZEPTEMBER 30.
Tapasztalataink Pompe-betegségben terhesség alatt alkalmazott enzimpótló kezeléssel és az irodalom áttekintése
A Pompe-betegség ritka, autoszomális recesszív módon öröklődő, izomdystrophiát okozó, lysosomalis tárolási betegség. Az α-glükozidáz enzim hiánya a sejtekben glikogénfelhalmozódást okoz. Az infantilis formában hypotonia, súlyos szív- és légzési elégtelenség, a késői kezdetű formában végtagövi és axiális eloszlású izomgyengeség, légzési elégtelenség tünetei jellemzők. A betegség 2006 óta a hiányzó enzim reguláris bevitelével kezelhető, ami mindkét altípusba tartozó betegek túlélését és tüneteinek súlyosságát szignifikáns mértékben javítja. A kezelés biztonságos és jól tolerálható, terhességben való alkalmazásáról azonban rendkívül kevés adat áll rendelkezésre. Célunk saját tapasztalataink megosztása és az irodalom áttekintése a Pompe-betegség enzimpótló kezelésének biztonságosságáról terhesség alatt és post partum.
Ideggyógyászati Szemle
2019. SZEPTEMBER 30.
Az epigenetikai szabályozás szerepe a kora gyermekkori betegségekben
Az 1990-es években a betegségek fejlődési eredete elmélet (Developmental origins of health and disease) térnyerésével elfogadottá vált, hogy a - DNS-szekvencia változásával nem járó - úgynevezett epigenetikai öröklődés szerepet játszik a betegségek kialakulásában.
Ideggyógyászati Szemle
2019. SZEPTEMBER 30.
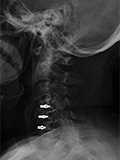
[1-es típusú neurofibromatosis, valamint duralis ectasia, vertebralis scalloping és csigolya-rendellenesség együttes előfordulása]
[A von Recklinghausen-kórként is ismert 1-es típusú neurofibromatosis (NF-1) gyakori autoszomális domináns kórkép, ami a lakosság körében 1:3000 arányban jelenik meg. Számos NF-1-beteg gerincdeformitások miatt kerül orvoshoz. Bőrgyógyászati ambulanciánkon egy 54 éves nőbeteg jelentkezett régóta fennálló, testszerte fokozatosan növekvő duzzanatok és foltok miatt. ]
Ideggyógyászati Szemle
2019. JANUÁR 30.
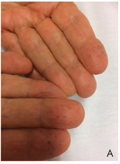
Multiplex ischaemiás stroke Osler-Rendu-Weber-kórban
A hereditaer haemorrhagiás teleangiectasia (HHT, Osler-Rendu-Weber-kór) olyan autoszomális domináns módon öröklődő, több lehetséges gén mutációja által okozott megbetegedés, amit az arteriovenosus rendszer több szervben is megjelenő malformációja jellemez. A klinikai diagnózist a Curaçao-kritériumok [1. spontán, visszatérő epistaxis; 2. karakterisztikus lokalizációban elhelyezkedő teleangiectasiák (ajak, szájüreg, orr, ujjak); 3. visceralis laesiók (gastrointestinalis, pulmonalis, cerebralis, spinalis); 4. elsőfokú érintett családtag] alapján állíthatjuk fel. A jelen közlemény célja Magyarországon elsőként egy multiplex ischaemiás stroke-kal társuló, genetikailag igazolt HHT-s eset ismertetése.
Hypertonia és Nephrologia
2018. DECEMBER 10.

Papillorenalis szindróma: a PAX2 békétlen mutációi
Az autoszomális domináns papillorenalis szindróma a PAX2 gén döntően de novo mutációinak következménye. A PAX2 egy transzkripciós faktort kódol, amely főként a vese, a húgyutak, az idegrendszer, a szem és a fül szöveteiben expresszálódik. Haploinsufficientiájának következtében a vese jellemzően hypoplasiás és hiperreflektív, de kialakulhat más fejlődési rendellenessége is. A klinikai megjelenést dominálhatja a nephroticus proteinuria, szövettanilag fokális szegmentális glomerulosclerosissal. Az okozott veseérintettség súlyossága rendkívül változó, a felismert esetek nagy részében az első négy évtizedben vezet végstádiumú veseelégtelenséghez. A PAX2-mutációk jellegzetes látóidegfő-eltérésekhez vezetnek, amelyek közül a leggyakoribb a látóidegfő (papilla) dysplasiája. A szindróma nevét meghazudtolva a PAX2-mutációt hordozó betegek negyedében nem alakul ki szemérintettség. Halláskárosodás ritkán, az esetek kevesebb mint 10%-ában társul. Az általunk azonosított öt családban az érintettek a másodiknegyedik évtizedben szorultak vesepótló kezelésre, teljes funkció vesztést okozó mutációk következtében.
Ideggyógyászati Szemle
2017. MÁRCIUS 30.
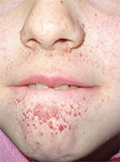
[Sclerosis tuberosás betegek gondozása]
[A sclerosis tuberosa egy autoszomális dominánsan öröklődő genetikai betegség, mely a legkülönbözőbb szervekben manifesztálódik benignus tumorok, illetve proliferációs zavarok formájában.]
Lege Artis Medicinae
2016. DECEMBER 18.
A családorvos feladatai a Wilson-kór miatt szükséges májtranszplantáció kapcsán
A Wilson-kór a 13-as kromoszómán lévő ATP7B gén mutációja következtében kialakuló, autoszomális recesszív módon öröklődő, rézfelhalmozódással járó betegség.
Ideggyógyászati Szemle
2016. JÚLIUS 30.
Transthyretin familiáris amyloid polyneuropathia - három magyarországi eset ritka mutációkkal (His88Arg és Phe33Leu)
A transthyretin familiáris amyloid polyneuropathia ritka, autoszomális domináns módon öröklődő progresszív szisztémás kórkép, amelynek patológiai háttere a transthyretin gén pontmutációja következtében kialakult endoneuralis amyloid depozíció.
1.
2.
3.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
4.
5.
1.
2.
Klinikai Onkológia
Hasnyálmirigyrák: az ESMO klinikai gyakorlati irányelve a diagnózishoz, kezeléshez, követéshez*
AUG 29.
3.
Klinikai Onkológia
Gyógyszerbiztonsági szemelvények – a múlt tanulságai és a jövő lehetőségei
AUG 29.
4.
5.