The eLitMed.hu medical portal uses computer cookies for convenient operation. Detailed information can be found in the Cookie-policy.
Clinical Neuroscience - 2008;61(11-12)
Content
Clinical Neuroscience
DECEMBER 20, 2008

[Pediatric intraventricular tumors]
[Pediatric intraventricular tumors present a well circumscribed group from surgical point of view. These tumors growing in the ventricular system cause hydrocephalus in most of the cases, the presenting symptoms are the signs of raised intracranial pressure. The mass lesion may remain silent for a long period, especially in infancy due to compensatory mechanisms, and the tumor might reach extreme size making the surgery a real challenge. This group has very specific postoperative problems resulting from the disturbance of CSF circulation. In this study we present the retrospective analysis of 55 patient operated for intraventricular tumor in the National Institute of Neurosurgery between 1991 and 2006. Data were analysed regarding histological type, presenting symptoms, type of surgical approach, radicalitiy of the resection and postoperative complications. In addition to our own results brief presentation of the specific histological groups is given based on the available literature.]



Clinical Neuroscience
DECEMBER 20, 2008

[Changes of the immune functions in patients with eating disorders]
[Aims - In this study we investigated whether calorie restriction or redundant food intake influences the function of regulatory T cells (Tregs), and their main regulators (dendritic cells and macrophages), or the targets of Tregs, CD4+ lymphocytes. Patients and methods - We investigated 11 white adolescents (10 girls and 1 boy) with anorexia nervosa, 12 obes adolescents and 10 healthy controlls. With flow cytometry we determined the prevalence of Tregs, myeloid and plasmacytoid dendritic cells. We applied intracellular staining to investigate TNF-alpha and IL-12 production of macrophages, moreover IL-2, IL-4, and IFN-gamma production of CD4+ cells. We also determined calcium flux kinetics upon activation in CD4+ cells. Results - We did not find any difference between obese, anorectic and control individuals in the prevalence of Tregs, dendritic cells, TNF-alpha and IL-12 positive macrophages, IL-4 and IFN-gamma positive CD4+ lymphocytes. We found that the prevalence of IL-2 positive lymphocytes after activation was lower in anorectic than in control subjects [median (range): 11.50 (7.60-15.30) vs. 13.50 (12.00-22.00), p=0.023], and in obese patients, too [12.50 (8.50-15.50) vs. 13.50 (12.00-22.00), p=0.028]. IFN-gamma/IL-4 ratio in CD4+ cells was higher in obese patients compared with control (p=0.046). The calcium flux characteristics of lymphocytes upon activation differed markedly in anorectic and healthy subjects as maximal calcium levels developed later in anorectic patients [86 (45- 232) vs. 215 (59-235) second, p<0.05]. We also tested the association between lymphocyte activation parameters and patients' clinical status, but did not find any association between the variables. Discussion - Our results suggest that the antigen presenting cell - regulatory T cell - CD4+ lymphocyte axis might be affected by calorie and nutritional disturbances, further studies are needed to elucidate the underlying processes.]
Clinical Neuroscience
DECEMBER 20, 2008
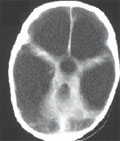
[Pneumococcal meningitis in children - 9 1/2-year-experience at Szent László hospital, Budapest, Hungary ]
[Background and objective - No recent publications are available about pneumococcal meningitis in Hungarian children. The aim of this study was to collect data of epidemiological, clinical and prognostic features of pneumococcal meningitis in children treated at Szent László Hospital, Budapest, Hungary. Methods - We conducted a retrospective review of medical charts and follow-up records of patients aged 1 to 18 years admitted to our Pediatric and Pediatric Intensive Care Units due to pneumococcal meningitis between 1st Jan 1998 and 30th Jun 2007. Results - 31 children with 34 cases of pneumococcal meningitis were admitted to our hospital in the study period. Two children developed recurrent illness. The mean age was 6 years, 26% were under 1 year of age. The mean duration of hospital stay was 21 days, 97% required intensive care. Frequent clinical symptoms were fever (100%), nuchal rigidity and vomiting (78%), altered mental status (71%), Kernig's and Brudzinski's signs (58%) and seizures (41%). Otitis media, sinusitis, mastoiditis were present in 44%, 58%, 41%, respectively. Subdural effusion, parenchymal cerebral lesion and sinus thrombosis were documented in 5, 3 and 2 cases, respectively. One third of the patients recieved ceftriaxon, two thirds were administered ceftriaxon and vancomycin. Adjunctive therapy with dexamethason was given to 91% of the children. 70% of patients required mechanical ventillation. 9 patients (25%) required endoscopic sinus surgery. In 13 cases (38%) mastoidectomy, in 5 children (15%) neurosurgery was performed. The case fatality rate was 23.5%. 8 (23.5%) patients had mild or moderate, 1 child (3%) developed severe neurological sequelae. Conclusion - Pneumococcal meningitis in children remains a source of substantial morbidity and mortality in childhood. The long hospital stay, the frequent need for intensive care and severe neurologic sequelae emphasize the importance of early diagnosis, early treatment and prevention with pneumococcal conjugate vaccines.]
Clinical Neuroscience
DECEMBER 20, 2008
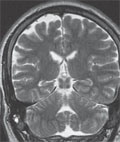
[Devastating epileptic encephalopathypseudoencephalitis: the new type of catastrophe epilepsy in our department]
[Purpose - Analysis of history of our five patients with intractable epilepsy whose illnes have begun with prolonged status epilepticus (SE) and high-grad fever of unknow cause. Methods - Retrospective study analysis of selected five intractable epileptic patients at a median age of 11.5 (8-14) years. Results - All children had normal development before epilepsy begun. Intractable SE lasted 3-10 (median seven) days by four patients and three months by one patient. The cause of illness was unknow at the beginning and the MRI were normal. Intractable epilepsy followed the SE in all cases without any latent period. Follow-up of the children was 3-15 (median 9.5) years. The seizures came continually with few-day-long breaks, antiepileptic drugs were ineffective. Semiology of seizures, EEG, and functional imaging examinations (PET, SPECT) referred to temporal and frontal lobe damages. Later on, the MR images showed hippocampal sclerosis in one patient and mild generalized brain atrophy in the others. During the years, cognitive deterioration and behavioral problems have been realized. The most severe patient developed tetraparesis, fell in vigil coma and died after five years. Conclusions - The symptoms of our patients fulfilled the criteria of devastating epileptic encephalopathy in schoolaged children.]
Clinical Neuroscience
DECEMBER 20, 2008
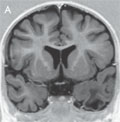
[How do temporal lobe seizures changeby age?]
[Seizure semiology describes different - motor, sensory, autonomic, etc. - aspects of epileptic seizures. Several semiological studies showed already that different epilepsies - especially temporal lobe epilepsy - contain age-dependent features. In our researches, we tried to assess these subjective aspects with as objective methods as possible. We gave a comprehensive (preictal, ictal, and postictal) description of seizure semiology in patients fulfilling the gold standard criteria of semiological studies: being seizure free after temporal lobe resections. Our studies based on a large population, assess epileptic features at different levels of brain maturation. They help to understand why certain semiological axes show special characteristics at different ages. In this review, I summarize the most important results of our seizure semiology studies in temporal lobe epilepsy.]
Clinical Neuroscience
DECEMBER 20, 2008
[Clinical and genetic diagnosis of dravet syndrome: report of 20 cases]
[Objective and background - Severe myoclonic epilepsy in infancy (SMEI; Dravet's syndrome) is a malignant epilepsy syndrome characterized by prolonged febrile hemiconvulsions or generalized seizures starting in the first year of life. Later on myoclonic, atypical absence, and complex partial seizures appear. When one of these seizure forms is lacking the syndrome of borderline SMEI (SMEB) is defined. Psychomotor delay resulting in mental retardation is observed during the second year of life. In most patients a de novo sodium channel alfa-1 subunit (SCN1A) mutation can be identified. By reviewing the clinical, laboratory, and neuroimaging data of our SMEI patients diagnosed between 2000 and 2008, we would like to share our experiences in this rare but challenging syndrome. Our results will facilitate the earlier and better diagnosis of Hungarian children with SMEI. Patients and methods - Clinical, EEG, MRI and DNA mutation data of 20 SMEI patients treated in the Bethesda Children’s Hospital (Budapest) were reviewed. Results - The first seizure appeared at age 6.3±3.0 months. At least one of the first two seizures were complex febrile seizures in 19/20 and unilateral seizures in 12/20 children. All children except for one showed hemiconvulsions at least once; all children had seizures lasting longer than 15 minutes. Eight of twenty patients had SMEB. DNA diagnostics identified an SCN1A mutation in 17 patients (6 missense, 4 nonsense, 4 frameshift, 2 splice site, 1 deletion) while 3 children had no mutation. Conclusion - Early diagnosis of SMEI is important for the avoiding unnecessary examinations and false therapies as well as for genetic counselling. Typical symptoms of SMEI are early and prolonged febrile hemiconvulsions with neurological symptoms, mental retardation and secondary seizure types later on. The presence of an SCN1A mutation supports the diagnosis. We propose the availability of molecular diagnostics and stiripentol therapy for SMEI children in Hungary.]
Clinical Neuroscience
DECEMBER 20, 2008

[Adrenocorticotrop hormon therapy in acquired childhood epileptic aphasia]
[Although Landau-Kleffner syndrome, a childhood-acquired epileptic aphasia, is frequently studied either the underlying pathophisiology or the optimal therapy remained unknown. In our study we aimed to investigate the efficacy of ACTH therapy in Landau-Kleffner syndrome. We have analysed retrospectively the documentation of five children treated by ACTH, who suffered from Landau-Kleffner syndrome. We studied the longitudinal changes of the four most characteristic sympthoms and signs of the syndrome: epileptiform EEG, speech and behaviour disorders, seizures together with the ACTH regimes. Besides, we analysed the relation between the starting date of the therapy and its efficacy. Before giving ACTH, epileptiform EEG and speech disorders were observed in all the five children, seizures in four of them, behaviour disorders in three of them. In two patients the speech disorder had been persisting for years before. Due to the starting ACTH stosstherapy (20 E/day for onetwo weeks) all the four examined signs disappeared or showed quick softening in all the five children in maximum two weeks. We adjusted long-term low dose maintenance therapy to avoid relapses in the long-term follow-up. Epileptiform EEGs have normalised in one case and have decreased in four cases. Speech disorders have disappeared in two and have softened in three children. Behaviour disorders have cured in 3/4 cases, softened in one case. Seizures have disappeared in all cases. One child is totally asymptomatic, four of them lives with softened symptoms. Analysing our data we found that the earlier the therapy starts, the more effective it is. On the basis of our data ACTH is an effective treatment for Landau-Kleffner syndrome. After giving it for only a short period, relapses often occur, to avoid relapses adjustment of long term low dose maintenance therapy is advisable.]
Clinical Neuroscience
DECEMBER 20, 2008
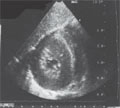
[Convulsions in neonatal period and infancy with rare etiology (neurogenetic disease)]
[Authors summerized the etiology of convulsions in neonatal period and infancy (hypoxy, intracranial hemorrhage, infections of central nervous system, metabolic background, chromosomal abnormalities, brain developmental abnormalities, benign neonatal convulsions, benign neonatal familial convulsions, drug withdrawal, inborn error of metabolism). They suggest screening examinations after convulsion, summerized the basic princpile of tandem examination and review a proposal at suspicion of inborn error of enzym defects (aminoacidemias, defects of fatty acid oxydation, organic acidemias). They present case history of two patients suffered in extraordinary inborn error of enzym defect (SCO2 gene mutation, propionic acidemia). Diagnosis originated in Heim Pál Hospital (settlement Madarász Hospital) with a Hungarian and international cooperation.]
Clinical Neuroscience
DECEMBER 20, 2008

[Hereditary neuropathy with liability to pressure palsy in childhood]
[HNPP is an autosomal-dominant inherited disease clinically characterized by painless, episodic, recurrent peripheral palsy often preceded by minor trauma or toxic damage. It generally develops during adolescence and rarely is reported in childhood. We observed two children with this disease. In one of the cases, also the child’s mother is suffering from HNPP. Clinical and genetic characterics of our three patients are summarized in this article.]
Clinical Neuroscience
DECEMBER 20, 2008

[Screening of hereditary neuromuscular disorders in the roma population living in Hungary]
[Recent medical genetic research has identified a number of novel, or previously known, but rare conditions, caused by private founder mutations. The Finnish and Ashkenazi Jew populations provide the best examples for identifying genes in unique genetic disorders. In these populations, research efforts and high-level medical services resulted in intense improvements of medical care and in organization of population- based screening programs. Hereditary disorders of the Roma populations are known for a long time. The genetic background of these diseases has been established by extensive molecular genetic studies. The Romas represent 6% of the Hungarian population and live under extremely bad health conditions. Therefore, our aim was to map the incidence of the hereditary neuromuscular disorders among the Hungarian Roma population. Moreover, we intended to provide proper information, genetic counseling and possible prevention strategies for the families at risk, which should represent a primer task in public health. Because of our experience in neuromuscular disorders, we choose six, frequent, autosomal recessive disorders for these clinical and genetic studies: hereditary motor and sensory neuropathy type Lom (HMSNL), hereditary motor and sensory neuropathy type Russe (HMSNR), congenital cataracts facial dysmorphism syndrome (CCFDN), Limb- Girdle muscular dystrophy 2C (LGMD2C), congenital myasthenic syndrome (CMS) and spinal muscular atrophy (SMA). Following identification of the founder mutations, the possibility of prenatal diagnosis and carrier screening for family members will contribute to the decrease of the recurrence risk for these severe, mostly untreatable disorders.]
1.
Clinical Neuroscience
[Headache registry in Szeged: Experiences regarding to migraine patients]
21. MAY
2.
Clinical Neuroscience
[The new target population of stroke awareness campaign: Kindergarten students ]
21. MAY
3.
Clinical Neuroscience
Is there any difference in mortality rates of atrial fibrillation detected before or after ischemic stroke?
27. NOV
4.
Clinical Neuroscience
Factors influencing the level of stigma in Parkinson’s disease in western Turkey
27. SEP
5.
Clinical Neuroscience
[The effects of demographic and clinical factors on the severity of poststroke aphasia]
18. JUL
1.
2.
Clinical Oncology
[Pancreatic cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up]
29. AUG
3.
Clinical Oncology
[Pharmacovigilance landscape – Lessons from the past and opportunities for future]
29. AUG
4.
5.