Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 6
Ideggyógyászati Szemle Proceedings
2023. OKTÓBER 26.

A Magyar Neuroimmunológiai Társaság X. Kongresszusa Absztraktfüzet 2.
A Magyar Neuroimmunológiai Társaság X. Kongresszusa Absztraktfüzet Siófok, Magyarország 2023. október 26–28.



Ideggyógyászati Szemle
2009. JANUÁR 20.

Genetikailag meghatározott neuromuscularis betegségek Magyarországon élõ roma családokban (angol nyelven)
A szerzők négy örökletesen meghatározott neuromuscularis kórkép klinikai és molekuláris genetikai aspektusait tenkintik át néhány magyarországi roma családban. Az autoszomális recesszíven öröklődő spinalis muscularis atrophia (SMA) csoportból nyolc, kaukázusi rasszhoz tartozó csecsemőben, kisdedben igazolták az SMA gén 7-8 exonalis deletióját, csupán két beteg tartozott a roma populációhoz. Hangsúlyozzuk, hogy a molekuláris genetikai lelet megegyezett a kaukázusi és roma populációban.

Magyar Radiológia
2008. MÁRCIUS 22.
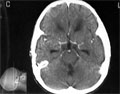
Egy gyermek sugárkezelésre felszívódó, rosszindulatú, intracranialis daganata
Our patient is a 9-year-old boy who was well until four months prior to admission to hospital following ataxia with persistent vomiting and later headaches. He was seen at the Sunyani Hospital in the northern part of Ghana and thereafter, referred to the neurosurgical department of the Korle-Bu Teaching Hospital, Accra. The patient, a class two pupil who lives with his parents, had no previous history of admission to hospital. On examination, he was ill-looking, though not pale, and afebrile. He was observed to be slightly wasted. He did not cough nor had dyspnoea. Chest radiograph was normal with uneventful cardiovascular system. He had ataxic gait and nystagmus, power was five in all limbs. Tone was slightly increased in the left lower limb. Laboratory data were nearly normal.
Ideggyógyászati Szemle
2005. JANUÁR 20.
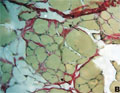
Végtagövi izomdisztrófiát okozó kalpaindefektus egy magyar családban
A végtagövi izomdisztrófiák (limb girdle, LGMD) csoportja klinikai és genetikai szempontból igen heterogén. A betegség fõ tünete a proximális végtagizmok progresszív gyengesége és atrophiája.
Ideggyógyászati Szemle
1997. NOVEMBER 20.

[Neurogenetikai betegségek - A diagnosztika és a terápia kérdései. Beszámoló az I. Neurogenetikai Tudományos Ülésről]
[Molekuláris genetikai alapfogalmak. A nem tradicionális öröklődéssel kapcsolatos alapfogalmak. A Southern-féle hibridizálás és a PCR-technika jelentősége a genetikai betegségek diagnosztikájában. A gén vizsgálata egy konkrét betegségben. Gyermekkori öröklődő idegrendszeri betegségek kivizsgálásának menete . Egy új transthyretin mutáció (TTRAsp18Gly) felderítését megnehezítő tényezőkről. A genetikai vizsgálatok szerepe a neurológiában. A genetikai laboratórium felépítése. A Magyarországon elvégezhető neurogenetikai vizsgálatok. A spinalis izomatrophia genetikai diagnosztizálásának újabb lehetőségei. A prenatalis vizsgálatok tapasztalatai. Trinukleotidrepeat-expanzió okozta neurológiai kórképek. A Huntington-betegség molekuláris genetikai diagnosztikája és annak problémái. Fragilis X-betegség molekuláris genetikai kimutatásának bevezetése hazánkban. A von Hippel-Lindau-betegség genetikai vonatkozásai. A molekuláris genetikai ismeretek fejlődése az öröklődő neuromuscularis betegségekben. A Charcot-Marie-Tooth 1. típusú perifériás neuropathia molekuláris genetikai diagnosztizálásának első hazai tapasztalatai. Heterozigóták vizsgálati problémái Duchenne-Becker-típusú myopathiákban. Analysis of IQ and genotype in Duchenne and Becker muscular dystrophy. Primer mitochondrialis myopathiák. Az mtDNS aberratióin alapuló nem primer mitochondriopathiák. A Leber-féle herediter opticus neuropathia magyarországi eseteinek feldolgozása során nyert tapasztalataink. Mitochondrialis deletiók detektálása PCR-módszerrel. Újabb adatok egyes epilepsziás tünetegyüttesek genetikai alapjaira. Új ismeretek az epilepszia genetikájában. Lysosomalis betegek osztályunk tíz éves beteganyagában. Biochemical diagnosis of inherited neuro-metabolic diseases. Neurogenetics in Germany. Prospect for gene therapy for the nervuous system and muscle. Preclinical research on gene therapy for Duchenne muscular dystrophy. Absence of apoe e4 allele in the neurofibrillary tangle subset of senile dementia. Mitochondrial DNA sequence variaton of complex I genes in Parkinson's disease. Ikervizsgálat epilepsziákban - Magyarországi multicentrikus vizsgálat. Az antiepileptikus hatás csökkenése terhes epilepsziás nőnél folsavtartalmú multivitamin adása mellett. Heredoataxiák molekuláris genetikai vizsgálata (SCAl, SCA2, SCA3, FA). Atrophia olivopontocerebellaris (Déjeriné-André-Thomas). A diagnosztika problémái. Hereditary Motor and Sensory Neuropathies (HMSN) in Southern Burgenland and Vicinity. Apolipoprotein E e4 allele frequency in patients with multiple system atrophy. Expression of nitric oxide synthase (NOS) isoforms in skeletal muscle of patients with mitochondrial. Pedigree analysis of hungarian Rett syndrome girls. Epidemiológiai vizsgálatok nyugat-dunántúli Huntington-kóros populációban. Pszichológiai tényezó'k szerepe Huntington-kór szempontjából veszélyeztetett egyének tanácsadása, illetve gondozása kapcsán. Lesch-Nyhan-szindróma . II. típusú spinalis izomatrophia klinikai elemzése. Familiaris scapuloperonealis szindróma és mitochondrialis DNS-defektus. Rilutek alkalmazása familiaris ALS-ben. Addison-kór és tünetek - adrenoleukodystrophia Esetismertetés. A familiaris hemiplegiás migrénről. Szomatikus mutációk vizsgálata kadáver agymintákon. A paramyotonia congenita EMG- és genetikai diagnosztikája. CTG-triplet-expanzió vizsgálata egy háromgenerációs myotonia dystrophiás családban.]
Ideggyógyászati Szemle
1995. MÁRCIUS 01.

THE PRINCIPLES OF GENE THERAPY FOR GENETIC DISEASES OF MUSCLE (DUCHENNE DYSTROPHY), SPINAL MOTOR NEURONS (SPINAL MUSCULAR ATROPHY) AND SCHWANN CELLS (SCHWANNOPATHIES)
Gene replacement therapy using adenoviral vectors in genetic diseases of the motor unit is a potential treatment modality. ln preclinical experiments, promising results have been obtained by dystrophin minigene replacement in mdx mice. There are several serious problems that remain to be resolved (i. e. immunologically mediated elimination of transduced cells) before this approach can be used in human clinical trials.
1.
2.
3.
4.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
5.
1.
2.
Klinikai Onkológia
A rosszindulatú daganatok fenotípusának plaszticitása és az immunogén mimikri
AUG 29.
3.
Klinikai Onkológia
A szarkopénia mérése komputertomográfiával és jelentősége az onkológiai betegeknél
AUG 29.
4.
5.