Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 345
Ideggyógyászati Szemle
2009. NOVEMBER 30.
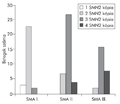
A spinalis izomatrophiát meghatározó survival motoneuron gének kvantitatív analízise
A spinalis izomatrophia (SMA) az egyik leggyakoribb autoszomális recesszíven öröklődő betegség, előfordulási gyakorisága 1/10 000, míg hordozósági frekvenciája közel 1/35. A betegséget a survival of motoneuron (SMN) ubiquiter fehérje hiánya okozza, amelyet a homológ SMN1 és SMN2 gének kódolnak.



Ideggyógyászati Szemle
2009. OKTÓBER 20.
Az öregedés fokozza az amiloid prekurzor protein és a triptofán 2,3-dioxigenáz gének transzkripcióját patkányagyban
A dementiák kialakulásának legfontosabb kockázati tényezője maga az öregedés. Hatvanöt éves kor után exponenciálisan nő az Alzheimer-kór (AK) gyakorisága, ötévente megduplázódik. Munkánk során azt vizsgáltuk, hogy az öregedés hogyan befolyásolja a neurodegeneratív kórállapotokban szerepet játszó gének transzkripcióját patkányagyban.
Lege Artis Medicinae
2009. SZEPTEMBER 21.

A D-vitamin-hiány jelentősége a gyakorlatban
A D-vitamin csonthatása már az 1920-as évek óta ismert. Az utóbbi években igazolódott, hogy szerepe a szervezetben jóval összetettebb. Az aktivált D-vitamin valójában szteroidhormon, amelynek receptora szinte minden sejttípusban kimutatható, és több mint 200 gén átíródásának szabályozásában van igazolt szerepe. Aktiválódása a régebbi elképzeléssel szemben nem csak a vesében lehetséges, sőt, a vázrendszeren kívüli hatásaiban a lokális 1-α-hidroxiláció fontosabb szerepet játszik.
Ideggyógyászati Szemle
2009. JÚNIUS 02.

A fájdalomküszöb eltérése szkizofréniában és állatmodellekben - II. rész
A fájdalomérzékenység csökkenése a szkizofrénia több mint 50 éve jól ismert tünete, azonban a hatásmechanizmusról nagyon kevés adat áll rendelkezésre, így ennek tisztázására megfelelő szkizofrénia-állatmodell szükséges. Két részben foglaljuk össze azokat az adatokat, amelyek a szkizofrénia és a fájdalomküszöb összefüggése vizsgálataiból származtak. A második fejezetben azokat a génkiütött állatokban létrehozott szkizofréniamodelleket tekintjük át, amelyekben a fájdalomküszöb vizsgálata megtörtént.
Lege Artis Medicinae
2009. MÁRCIUS 21.
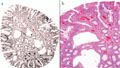
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.
Ideggyógyászati Szemle
2009. JANUÁR 20.

Genetikailag meghatározott neuromuscularis betegségek Magyarországon élõ roma családokban (angol nyelven)
A szerzők négy örökletesen meghatározott neuromuscularis kórkép klinikai és molekuláris genetikai aspektusait tenkintik át néhány magyarországi roma családban. Az autoszomális recesszíven öröklődő spinalis muscularis atrophia (SMA) csoportból nyolc, kaukázusi rasszhoz tartozó csecsemőben, kisdedben igazolták az SMA gén 7-8 exonalis deletióját, csupán két beteg tartozott a roma populációhoz. Hangsúlyozzuk, hogy a molekuláris genetikai lelet megegyezett a kaukázusi és roma populációban.
Ideggyógyászati Szemle
2008. DECEMBER 20.
A dravet-szindróma klinikai és genetikai diagnosztikájáról húsz esetünk kapcsán
A Dravet-szindrómát vagy más néven súlyos csecsemőkori myoclonus epilepsziát az addig minden szempontból jól fejlődő csecsemő esetében az első életévben fellépő, eleinte többnyire lázas, majd láztalan, elhúzódó, generalizált vagy féloldali, általában változó oldali, clonusos vagy tónusos-clonusos rohamok jellemzik, amelyekhez később myoclonusok, atípusos absence-ok, komplex parciális rohamok társulnak.
Ideggyógyászati Szemle
2008. DECEMBER 20.

Herediter nyomásérzékeny neuropathia gyermekkorban
A herediter nyomásérzékeny neuropathia autoszomális domináns öröklődésű kórkép, amit klinikailag rekurráló perifériás paresisek jellemeznek többnyire benignus lefolyással. Kiváltó okként gyakori a minor trauma vagy toxikus behatás. A perifériás ideg szövettani képe alapján „tomaculous” neuropathiának is nevezik, mivel a szegmentális de- és remyelinisatio a perifériás idegen hurkafűzérszerű képet okoz.
Ideggyógyászati Szemle
2008. JÚLIUS 30.
Az emberi agy és intelligencia evolúciója
Az élővilág és benne az ember evolúciójának fő hajtóereje a sikeres alkalmazkodás a környezeti változásokhoz, amit véletlenszerű genetikai módosulások és az ebből fakadó „újítások” tesznek lehetővé. Jelenlegi tudásunk szerint az emberré válás körülbelül 7-8 millió évvel ezelőtt az afrikai szavannákon kezdődött el, ahol a felegyenesedett testtartás és a két lábon járás jelentős evolúciós előnyökkel járt.
Lege Artis Medicinae
2008. MÁJUS 26.
A juvenilis polyposis szindróma igazi arca
BEVEZETÉS - A colorectalis tumorok javarészt sporadikusan fordulnak elő, de számolni kell a familiáris halmozódású, illetve autoszomális domináns öröklődésű kórképekkel is. A juvenilis polyposis szindróma autoszomális, domináns öröklődésű kórkép, amelynek hátterében az SMAD4 vagy a BMPR1A gén mutációja állhat. A kórképre a hamartomatosus polipok nagy száma jellemző, amelyek megtalálhatók mind a felső gastrointestinumban, mind pedig a colorectum területén. A korábbi vélelmek ellenére a polipok egy része, átlagosan a 34-35. életév táján malignus átalakuláson mehet keresztül, ahogy az általunk követett család esete is mutatja. ESETISMERTETÉS - A juvenilis polyposis miatt kezelt fiatalember 18 éven át tartó, folyamatos gondozása során több mint száz polip került eltávolításra a gastrointestinumból. Nyolcéves gondozási szünet után (elégtelen compliance) betegünk 31 éves korában, súlyos klinikai állapotban érkezett vizsgálatra, amelynek során metasztatizáló colorectalis carcinomát észleltünk. A beteg rövid palliatív terápia után elhunyt. A beteg családfája alapján vizsgáltuk élő, nagykorú, elsőfokú rokonait, akik közül testvérbátyjánál szintén igazolódott a juvenilis polyposis szindróma. Az elvégzett genetikai vizsgálatok a BMPR1A mutációját igazolták a klinikailag is beteg testvérnél, annak egyik gyermekénél és az elhunyt gyermekénél is. KÖVETKEZTETÉS - A genetikai vizsgálatokkal a mutációt nem hordozókat mentesíthettük a betegségtudat nyomasztó terhe és nem utolsósorban a felesleges klinikai, főleg invazív vizsgálatok alól, míg a mutációt hordozókat a lehető legnagyobb klinikai gondossággal tudjuk ellenőrizni.
1.
2.
3.
4.
5.
Egészségpolitika
Hadiállapotként kezeli és így is reagál a kormány az egészségügy „rendezésére”
2024 JÚN 18.
1.
2.
3.
Mesterséges intelligencia
A legújabb, MI által asszisztált robotsebészet megérkezett Magyarországra
2024 SZEPT 27.
4.
Orvoslás és társadalom
Különleges nap halmozottan sérült gyermekeket nevelő szülők számára a Bükki Nemzeti Parkban
2024 SZEPT 27.
5.