Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.
Részletes keresés
Kérjük, állítsa be a paramétereket!
Találatok száma: 26
Ideggyógyászati Szemle
2011. NOVEMBER 30.
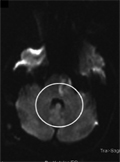
A mitochondrialis DNS A8344G-mutációja típusos mitochondrialis encephalomyopathia laktátacidózissal és stroke-szerű epizódokkal járó szindrómában
Juvenilis ischaemiás stroke szindróma ritka esetét mutatjuk be, amelyben a mitochondrialis DNS A8344G mutációja igazolódott a tRNSLys génben. A beteg klinikai fenotípusa jellegzetes MELAS szindróma (mitochondrialis encephalomyopathia laktátacidózissal és stroke-szerű tünetekkel) volt.



Lege Artis Medicinae
2011. FEBRUÁR 20.

Koenzim-Q10, rosuvastatin és klinikai eredmények szívelégtelenségben
A koenzim-Q10 (ubiquinon) a természetben előforduló, zsíroldékony quinon, amely elektrontranszporterként hatva a mitochondrialis oxidatív foszforiláció és az adenozin- trifoszfát (ATP) képzésének esszenciális kofaktora. A koenzim-Q10 redukált formájában valószínűleg lipofil antioxidánsként is hat, védi a sejtmembránokat és a keringő lipoproteineket az oxidációtól.
Ideggyógyászati Szemle
2009. JÚNIUS 02.

99-mTc-HMPAO egyesfoton-emissziós komputertomográfia genetikailag meghatározott neurometabolikus betegségekben
A vizsgálat célja a regionális cerebralis véráramlás rendellenességeinek a meghatározása volt enzymopathiák különböző típusaiban. Betegek és módszerek - A genetikailag meghatározott enzymopathiában szenvedő betegek közül három esetben volt jelen aminoacidopathia, 11 beteg szenvedett különböző típusú encephalopathiában, közülük tíz mitochondrialis encephalopathiában (MEMP), egy esetben volt jelen hyperuricaemiás encephalopathia. A felsorolt 14 beteg mellett még egy beteg ceroid lipofuscinosisban, egy másik pedig scleoris tuberosában szenvedett.
Magyar Immunológia
2008. JANUÁR 22.
Sejtélettani folyamatok jellemzése valós idejű áramlási citometriás módszerrel
Az áramlási citométerek számos sejtpopulációban teszik lehetővé több sejtélettani folyamat egyidejű (real-time) monitorozását. A vizsgálatok során a sejtbe olyan festéket juttatnak, ami gerjesztés hatására fluoreszkál, és az általa kibocsátott fluoreszcens jel érzékenyen követi a vizsgálandó paraméter változását.
Ideggyógyászati Szemle
2003. AUGUSZTUS 20.

Apoptózis fokális agyi ischaemiában
Az ischaemiás stroke, amely hazánkban is vezető halálok, gócos agyi vérellátási zavar következtében alakul ki. Az ischaemiás sejtpusztulás döntően hevenyen lezajló nekrózis formájában jelentkezik. Terápiás szempontból is jelentős sajátosság, hogy a nekrotikus agyterület határzónájában késleltetett sejtvesztés zajlik, amely az apoptózis jellegzetességeit mutatja. Az apoptózis korunk orvosbiológiai kutatásainak egyik legintenzívebben vizsgált folyamata, mechanizmusának jobb megismerése számos betegség, köztük az ischaemiás stroke eredményesebb terápiáját ígéri.
Ideggyógyászati Szemle
2001. MÁJUS 01.

Elektroneurográfiás és elektromiográfiás eltérések mitochondrialis megbetegedésekben
A mitochondrialis kórképekben a betegség leggyakrabban a posztmitotikus szövetekben manifesztálódik, így a klinikai tünetek a központi és a környéki idegrendszer, a váz-, illetve szívizom működészavarai, az endokrin szervek rendellenességei vagy hepatopathia formájában jelentkeznek.
Ideggyógyászati Szemle
2001. MÁRCIUS 01.

A migrén genetikája. Összefoglaló tanulmány
Az elmúlt évtizedben jelentősen gyarapodtak ismereteink a migrén öröklődéséről. Populációgenetikai vizsgálatok eredményei valószínűsítik, hogy a migrén poligénesen öröklődő betegség.
Ideggyógyászati Szemle
2000. NOVEMBER 01.

[Mitochondrialis enzimaktivitásvizsgálatok különböző típusú mitochondrialis myopathiákban/encephalopathiákban]
[A szerzők mitochondrialis oxidatív foszforilációs (OXYPHOS) enzimrendszer [NADH-citokrómoxidoreduktáz (NADH-COR: respiratorikus enzim komplex 1), citokróm-c-oxidáz (COX, IV respiratorikus enzimkomplex), karnitin-acetil-transzferáz, citrát-szintáz, lipoamid-dehidrogenáz] aktivitását vizsgálták izombiopsziás anyagból 12, szövettanilag igazoltan mitochondrialis myopathiás/ encephalomyopathiás egyén esetében: 11 gyermek és 1 felnőtt beteg. Nyolc betegnél COX-hiány, négynél NADH-COR-enzimhiány igazolódott. Három familiáris ataxiás gyermek esetében csökkent karnitin-acetil-transzferáz-aktivitás társult a COX-defektushoz, további három esetben csökkent lipoamid-dehidrogenáz-aktivitás volt detektálható. Klinikai típus szerint egy neonatalis, hét infantilis és négy adult eset szerepelt. Az izombiopszia hisztokémiailag diffúz gyenge vagy gócokban hiányzó COX-aktivitást és atrophiát bizonyított. Három gyermeknél találtak úgynevezett tépett vörös rostokat módosított Gömöri-festéssel.]
Lege Artis Medicinae
2000. SZEPTEMBER 01.

Ideggyógyászati Szemle
2000. JÚLIUS 01.

[Súlyos/fatális infantilis citokróm c-oxidáz-hiány (biokémiai és morfológiai vizsgálatok)]
[A szerzők négy, súlyos állapotú csecsemő esetét közlik, akik generalizált izomhypotoniában, cardiorespiratoricus elégtelenségben, súlyos izomatrophiában, citokróm c-oxidáz (COX) -defektusban szenvedtek. lzombiopsziából nyert mintában a mitochondrialis enzimek aktivitását vizsgálták: citokróm c-oxidáz, citrátszintáz, malátdehidrogenáz, karnitin-acil-transzferáz. Mind a négy esetben COX-defektust igazoltak, laktátacidosissal és/vagy hyperpyruvataemiával. Egy esetben a szövettani lelet mitochondrialis myopathiának felelt meg, három esetben azonban Werdnig-Hoffmann-szerű neurogén izomatrophiának. Két csecsemőt kellett gépen lélegeztetni életük utolsó heteiben. Súlyos mitochondrialis COX-hiányban hypotoniás, laktátacidosisos és/vagy hyperpyruvataemiás újszülöttek/ csecsemők esetében elkülönítő kórismeként mitochondrialis enzimdefektusra, így COX-hiányra is gondolni kell, a piruvát-karboxiláz, a piruvát-dehidrogenáz és a zsírsavak mitochondrialis,β-oxidációs defektusának kizárása után.]
1.
2.
3.
Ideggyógyászati Szemle Proceedings
Egészségügyi szakmai irányelv az akut ischaemiás stroke diagnosztikájáról és kezeléséről
AUG 31.
4.
5.
1.
2.
Klinikai Onkológia
A rosszindulatú daganatok fenotípusának plaszticitása és az immunogén mimikri
AUG 29.
3.
Klinikai Onkológia
A szarkopénia mérése komputertomográfiával és jelentősége az onkológiai betegeknél
AUG 29.
4.
5.