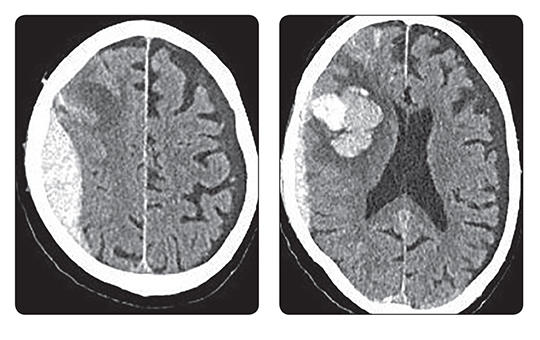
A blokkolóórák után most már személyesen ellenőrök is figyelik, ki tartózkodik a kórházakban. Ki dolgozik, ki fekszik a kórházi ágyakon, és hogyan használják például a diagnosztikai képalkotókat. Bármely nap és a nap bármely órájában érkezhet a mobil egység. Figyelnek, ellenőrzik, leleplezik, „lógnak”-e az egészségügyben dolgozók.
Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.