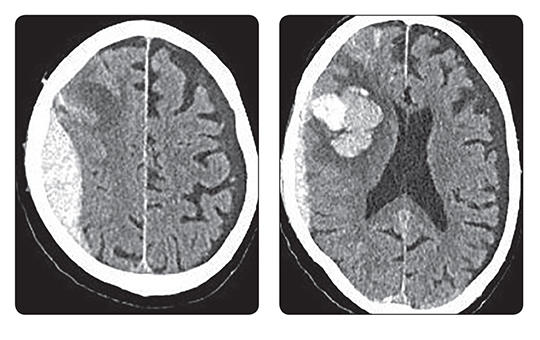
Soha nem látott ütemben nőnek a kórházi adósságok. Úgy tűnik, a béremelés még jobban felgyorsította a folyamatot: míg 2023-ban átlagosan havonta 3,7 milliárddal nőtt ez az adósság, 2024-ben havi 15-20 milliárddal, ami már a működésüket veszélyezteti. Inkasszózik a NAV, de már a beszállítók is.
Az eLitMed.hu orvostudományi portál a böngészés tökéletesítése érdekében cookie-kat használ.
Ha bővebb információkat szeretne kapni a cookie-k használatáról és arról, hogyan módosíthatja a beállításokat, kattintson ide: Tájékoztató az eLitMed.hu Cookie-használatáról.